In order to explain these patterns, it was sometimes argued that some bacteria have small genomes because there is selection for rapid cell division, with larger DNA contents taking longer to replicate and thereby slowing down the cell cycle. However, when Mira et al. (2001) compared doubling time and genome size in bacteria that could be cultured in the lab, they found no significant relationship between them. In other words, selection for small genome size is probably not responsible for the highly compact genomes of some bacteria, even though it seems plausible that, more generally, selection does prevent the accumulation of non-coding DNA to eukaryote levels in bacterial cells.
Mira et al. (2001) suggested a different interpretation that is based on two other major processes in evolution — mutation and genetic drift. In terms of mutation, they pointed out that on the level of individual changes that add or subtract relatively small quantities of DNA — i.e., insertions or deletions, or “indels” — deletions tend to be somewhat larger than insertions. The insertions in this case are separate from the addition of whole genes, which happens often in bacteria through sharing of genes among individuals or even across species (“horizontal gene transfer” or “lateral gene transfer“) or gene duplication.
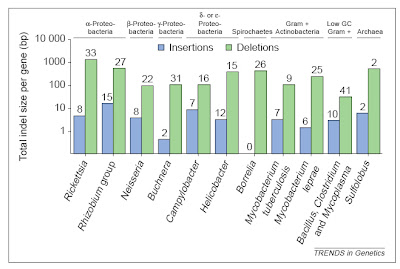 In bacteria (and eukaryotes) small-scale deletions tendto involve more base pairs than insertions, creating a“deletion bias”. Of course, larger insertions such as oftransposable elements or gene duplicates are not partof this calculation as they add much more DNA at once. From Mira et al. (2001).
In bacteria (and eukaryotes) small-scale deletions tendto involve more base pairs than insertions, creating a“deletion bias”. Of course, larger insertions such as oftransposable elements or gene duplicates are not partof this calculation as they add much more DNA at once. From Mira et al. (2001).So, on the one hand, there are processes that can add genes (duplication and lateral gene transfer), whereas in the absence of these processes, and if there are no adverse consequences to losing DNA (i.e., there is no selective constraint occurring), genomes should tend to get smaller as a result of this deletion bias. In free-living bacteria, there are many opportunities for gene exchange, with lateral gene transfer adding DNA at an appreciable frequency. Moreover, free-living bacteria tend to occur in astronomical numbers, and elementary population genetics reveals that selection will be strong under such conditions (so that even a mildly deleterious mutation, such as a deletion or disruptive insertion, will probably be lost from the population over time). Finally, free-living bacteria must produce their own protein products, and therefore tend to make use of all their genes, which places selective constraints on changes (including indels) in those sequences.
Endosymbiotic bacteria, especially those that live within the cells of eukaryote hosts, are different in multiple relevant respects. First, they do not regularly encounter other bacteria from whom they can receive genes. Second, they occur in drastically smaller numbers — indeed, they experience a population bottleneck severe enough to shift the balance from selection to drift. Third, they come to rely on some metabolites provided by the host and no longer make use of all their own genes. These factors in combination mean that the selective constraints on many endosymbiont genes are relaxed, and the dominant processes become deletion bias and random drift. Over many generations, endosymbiotic bacteria lose the genes they are not using (and some that are only mildly constrained by selection, such is the strength of drift under such conditions) due to deletion bias, and the end result is highly compact genomes.
The compaction of genomes in endosymbionts can be extreme. The smallest genome known in any cellular organism (except, perhaps, one in Craig Venter‘s lab) is found in the bacterial genus Carsonella, a symbiont that lives within the cells of psyllid insects. It contains only 159,662 base pairs of DNA and 182 genes, some of which overlap (Nakabachi et al. 2006).
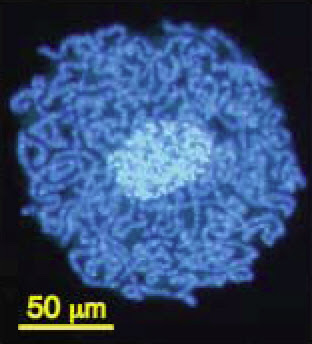 Carsonella (dark blue) living within the cells and
Carsonella (dark blue) living within the cells and
around the nucleus (light blue) of a psyllid insect.
From Nakabachi et al. (2006).In some other bacteria, genes that are not used (including non-functional duplicates) may not be lost for some time and may persist as pseudogenes, just as are observed in large numbers in eukaryote genomes. These tend to undergo additional mutations and to degrade over time but can still be recognized as copies of existing genes. In Mycobacterium leprae, the pathogen that causes leprosy, for example, there are more than 1,100 pseudogenes alongside roughly 1,600 functional genes (Cole et al. 2001). Its genome is about 1 million base pairs smaller than that of its relative M. tuberculosis, but clearly many of the inactive genes have not (yet) been deleted.
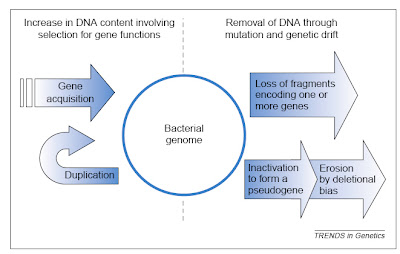 The two major influences on bacterial genomes: insertion ofgenes by duplication and lateral gene transfer, and the lossof non-functional sequences by deletion.From Mira et al. (2001).
The two major influences on bacterial genomes: insertion ofgenes by duplication and lateral gene transfer, and the lossof non-functional sequences by deletion.From Mira et al. (2001).It would be nice if this post could end there, having delivered a brief overview of an interesting issue in comparative genomics. Sadly, there is more to say because some anti-evolutionists apparently have begun using the topic in a confused attempt to challenge evolutionary science. In particular, though I note that I have become aware of this only second hand, some creationists apparently have suggested that all bacterial genomes are degrading and therefore that bacteria today are simpler than they were in the past, such that complex structures like flagella could not have evolved from less complicated antecedents.
It should be obvious that not all genomes are necessarily “degrading” just because there is a net deletion bias. For starters, selective constraints prevent essential genes from being lost by this mechanism in most bacteria. Furthermore, there exist well established mechanisms that can add new genes to bacterial genomes, including lateral gene transfer and gene duplication. In fact, the rate of gene duplication seems to be related to genome size in bacteria (Gevers et al. 2004). Also, as Nancy Moran noted in an email, “The most primitive bacteria were certainly simple, but they are not around or at least are not easily identified. Many modern bacteria have large genomes and are very complex.” Finally, the compact genomes of endosymbionts, such as in the aphid symbiont Buchnera aphidicola, tend to be more stable than the genomes of free-living bacteria in terms of larger-scale perturbations such as chromosomal rearrangements (Silva et al. 2003).
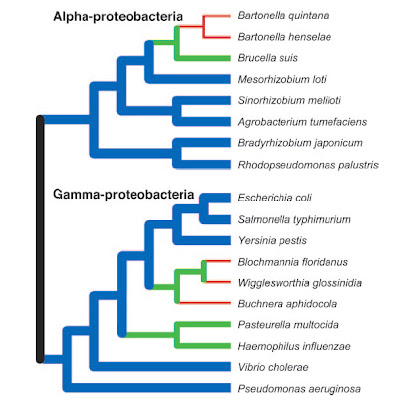 Some bacteria, in particular those that have shifted to a
Some bacteria, in particular those that have shifted to a
parasitic or endosymbiotic dependence on a eukaryote host,
have undergone genome reductions (green, red) as compared
to inferred ancestral conditions. Nevertheless, many other
species continue to display large genomes (blue).
However, the very earliest bacteria probably began
with small genomes and simple cellular features.
From Ochman (2006).As with eukaryotes, the genomes of bacteria provide exceptional confirmation of the fact of common descent. Not only do comparative gene sequence analyses shed light on the relatedness of different bacterial lineages and the evolution of features like flagella, but the presence — and loss to varying degrees — of non-functional DNA highlights a strong historical signal.
Given that it is her work that is being misused by anti-evolutionists, it is fitting that Dr. Moran be given the last word:
“It seems to me that the widespread occurrence of degrading genes, which are present in most genomes including those of animals, plants, and bacteria, argues pretty strongly in favor of evolution. They are the molecular equivalent of vestigial organs.”
Quite right.
_____________
References
Cole, S.T., K. Eiglmeier, J. Parkhill, K.D. James, N.R. Thomson, P.R. Wheeler, and et al. 2001. Massive gene decay in the leprosy bacillus. Nature 409: 1007-1011.
Gevers, D., K. Vandepoele, C. Simillion, and Y. Van de Peer. 2004. Gene duplication and biased functional retention of paralogs in bacterial genomes. Trends in Microbiology 12: 148-154.
Gregory, T.R. 2005. Synergy between sequence and size in large-scale genomics. Nature Reviews Genetics 6: 699-708.
Gregory, T.R. and R. DeSalle. 2005. Comparative genomics in prokaryotes. In The Evolution of the Genome, ed. T.R. Gregory. Elsevier, San Diego, pp. 585-675.
Mira, A., H. Ochman, and N.A. Moran. 2001. Deletional bias and the evolution of bacterial genomes. Trends in Genetics 17: 589-596.
Nakabachi, A., A. Yamashita, H. Toh, H. Ishikawa, H.E. Dunbar, N.A. Moran, and M. Hattori. 2006. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science 314: 267.
Ochman, H. 2006. Genomes on the shrink. Proceedings of the National Academy of Sciences of the USA 102: 11959-11960.
Ochman, H. and L.M. Davalos. 2006. The nature and dynamics of bacterial genomes. Science 311: 1730-1733.
Silva, F.J., A. Latorre, and A. Moya. 2003. Why are the genomes of endosymbiotic bacteria so stable? Trends in Genetics 19: 176-180.








 319,000 human ERV LTR regions have a near-perfect p53 DNA binding site. The LTR10 and MER61 families are particularly enriched for copies with a p53 site. These ERV families are primate-specific and transposed actively near the time when the New World and Old World monkey lineages split. Other mammalian species lack these p53 response elements. Analysis of published genomewide ChIP data for p53 indicates that more than one-third of identified p53 binding sites are accounted for by ERV copies with a p53 site. ChIP and expression studies for individual genes indicate that human ERV p53 sites are likely part of the p53 transcriptional program and direct regulation of p53 target genes. These results demonstrate how retroelements can significantly shape the regulatory network of a transcription factor in a species-specific manner.
319,000 human ERV LTR regions have a near-perfect p53 DNA binding site. The LTR10 and MER61 families are particularly enriched for copies with a p53 site. These ERV families are primate-specific and transposed actively near the time when the New World and Old World monkey lineages split. Other mammalian species lack these p53 response elements. Analysis of published genomewide ChIP data for p53 indicates that more than one-third of identified p53 binding sites are accounted for by ERV copies with a p53 site. ChIP and expression studies for individual genes indicate that human ERV p53 sites are likely part of the p53 transcriptional program and direct regulation of p53 target genes. These results demonstrate how retroelements can significantly shape the regulatory network of a transcription factor in a species-specific manner.{kind=link}