Sometimes it is helpful to have a catchy word to describe one’s type of research. I think that’s why “omics” words are so popular — they encapsulate a complex combination of approaches (usually something + genomics, or something-more-than-genomics) in a memorable way that immediately conveys the gist of the field. “Metagenomics” is a good example — it’s the study of a larger assemblage of genomes than just one, usually from an environmental sample of microbes. “Proteiomics” is another, or “transcriptomics”. Of course, this can get out of hand (see here). However, I think the study of genome size (which predates molecular genetics, let alone genomics) deserves a catchy moniker. The problem is, I haven’t really come up with one in the past, so I just end up saying “I study the total amount of DNA in different species of animals, which includes genes and all the other sequences, most of which are non-coding and…” — well, you get the idea.
People like me study entire genomes — every component included, be it gene or pseudogene or repeat or transposable element. We also are interested, not in a few model organisms, but in everything (people usually stare blankly when, to the question “which animals do you work on?”, I reply “all of them”). But what to call such a discipline?
Proposed neologism: “Omnigenomics”
Etymology: Latin “omnis” (all or everything) + genomics (study of genomes)
Sample usage: “What do you do?” / “Omnigenomics” / “What’s that?” / “I study the total amount of DNA in different species of animals, which includes genes and all the other sequences, most of which are non-coding and…”
(The alternative Greek version, “pangenomics”, is already being used and sounds way less cool).
(Yes, I note the irony of getting a “worst new omics word award” from a blog called phylogenomics.blogspot.com!)
Wilhelm Johannsen, who coined such terms as “gene”, “genotype”, and “phenotype”, noted in 1911 that,
It is a well-established fact that language is not only our servant, when we wish to express – or even to conceal – our thoughts, but that it may also be our master, overpowering us by means of the notions attached to the current words.
Even widely used and (apparently) simple terms can cause substantial confusion when the notions attached to them are unwarranted, a problem that is particularly common in evolutionary biology. Think “theory“, a term that is not only poorly understood to begin with in its scientific context but is actively misrepresented by anti-evolutionists.
The misinterpretation of other terms may cause, or be caused by, particular assumptions about the nature of the evolutionary process. Take the term “primitive” for example. The use of the term can be either deeply misleading or entirely appropriate depending on what one considers to be the opposite of the term. Here are the two most common uses, the first problematic and the second legitimate.
1) Primitive versus advanced. In this comparison, the term “primitive” has pejorative connotations of inferiority relative to “more evolved” species. Evolution is implied to be a progressive process characterized by improvement rather than simply of change. This is the usage one finds in popular media, for example in the all-too-well-known “evolutionary line-up” showing progressive improvement in anything from primate species to any manner of product being advertised as new and improved. However, biological evolution is not a progressive process, and this use of the term is inappropriate.
2) Primitive versus derived. In technical parlance, “primitive” can be used to mean that one form of a trait is “more like a common ancestor” relative to another form of the trait (i.e., as synonymous with “ancestral”). It is an expression of the differential quantity of change that has occurred since two or more lineages diverged. The opposite of primitive in this usage is not “advanced” but “derived”. There is no automatic implication that change has been progressive in this sense.
So, one should not draw a comparison between “primitive versus advanced”, but “primitive versus derived” is not problematic. It bears noting, though, that the terms primitive (or ancestral) and derived are actually applicable to particular characters, not to entire organisms. As Crisp and Cook (2005) noted,
Once two lineages have separated, each evolves new characters independently of the other and, with time, each will show a mixture of plesiomorphic [inherited largely unchanged from the ancestor] and apomorphic [newly evolved and thus not possessed by the ancestor] character states. Therefore, extant species in both lineages resemble, to varying degrees, their common ancestor. Consequently, whereas character states can be relatively ancestral (plesiomorphic) or derived (apomorphic), these concepts are nonsensical when applied to whole organisms.
_______
Crisp, M.D. and L.G. Cook. 2005. Do early branching lineages signify ancestral traits? Trends in Ecology and Evolution 20: 122-128.
Johannsen, W. 1911. The genotype conception of heredity. American Naturalist 45: 129-159.
As I and others have noted many times, facts, theories, and hypotheses are independent elements in the scientific process. Contrary to their vernacular meanings, they are not ranks indicating differential degrees of certainty in some claim.
Evolution is scientific fact, meaning that the numerous types of evidence point so overwhelmingly to shared ancestry that scientists have accepted it as true about the world, in the same provisional-but-extremely-likely sense that they accept other facts like gravity or the existence of atoms.
Evolution is also a theory, meaning that there is a cohesive body of mechanistic explanations that seeks to explain the historical fact of common descent. This includes, but is not limited to, random processes like genetic drift, quasi-random ones such as mutation, and absolutely non-random ones like natural selection.
In addition to being a fact (that species are related through common ancestry) and a theory (well supported mechanisms that explain how evolutionary change happens), evolution represents the unique historical path that living lineages have followed. Whereas there is no longer any real disagreement in biology (or indeed, in science in general) over the historical factuality of common relatedness, evolutionary biology is rife with heated debate regarding the mechanisms and their relative importance, the specific historical relationships linking related groups, and the intermediate steps that occurred in the origin of particular features. As such, hypotheses are also important in evolutionary biology, because they represent testable statements that are used to support or refute specific details of theory or path.
One of the more stinging criticisms that evolutionary biologists level at each other when they argue about evolution as path is to call a proposed account a “just-so story”. The phrase itself comes from Rudyard Kipling’s Just So Stories from the early 1900s, many of which included fantastical accounts of the origins of particular features, like the elephant’s trunk, the leopard’s spots, and the giraffe’s neck. According to Wikipedia,
A just-so story, also called the ad hoc fallacy, is a term used in academic anthropology, biological sciences, and social sciences. It describes an unverifiable and unfalsifiable narrative explanation for a cultural practice or a biological trait or behavior of humans or other animals. The use of the term is an implicit criticism that reminds the hearer of the essentially fictional and unprovable nature of such an explanation. Such tales are common in folklore and mythology.
The important criterion in whether something represents a just-so story rather than a hypothesis is a lack of testability and hence unfalsifiability. A lack of complete supporting data in itself is not an indicator that something is a just-so story, because many hypotheses also lack these prior to being subjected to testing. In other words, what matters is whether it can be tested, not whether it has already been tested, though obviously some supporting data must be provided eventually if the hypothesis is to be considered seriously.
Creationists, who seem unequipped with irony detectors, tend to dismiss all hypotheses about evolutionary path, even ones for which there is substantial supporting evidence, as just-so stories. In fact, they often demand an absurd amount of evidence and detail, such as an observable, repeatable, mutation-by-mutation demonstration of some feature evolving. When this obviously cannot be delivered (nor could its counterpart in any science), they believe this supports their unobservable, unrepeatable, vague explanation for the feature’s origins. Theirs is the ultimate just-so story, but that does not prevent them from projecting onto scientists.
As a case in point, consider the infamous bombardier beetles, which I mentioned briefly in a recent post. Along with eyes, blood clotting, the immune system, and bacterial flagella, the defence mechanism of these intriguing beetles supposedly represents an un-evolvable feature due to its irreducible complexity.
The claims made by creationists about these beetles relate to both fact and path. In terms of fact, they often suggest that the reagents used in its defensive system will explode when mixed together, and thus that their mechanism of storing them separately must have arisen fully formed. This is demonstrably false. A catalyst is required, as Dawkins showed by mixing the liquids.
With regard to path, they argue that a series of functional intermediates in the gradual evolution of the defensive apparatus is impossible in principle. This claim is also easily refuted by the presentation of plausible, testable hypotheses showing how functional intermediates could have occurred. One example is given in this video.
Now, is this the answer to how the spectacular defence system of bombardier beetles arose? I do not know, but I suspect probably not. A similar point of view is presented by Mark Isaak, who authored the Talk.Origins article Bombardier beetles and the argument of design:
The scenario above is hypothetical; the actual evolution of bombardier beetles probably did not happen exactly like that … Determining the actual sequence of development would require a great deal more research into the genetics, comparative anatomy, and paleontology of beetles. The scenario does show, however, that the evolution of a complex structure is far from impossible. The existence of alternative scenarios only strengthens that conclusion.
Are such scenarios just-so stories? No, they are hypotheses, and they are testable. For example, are the hypothetical intermediate stages found to be functional in any other species? Are the chemicals used in combination in the defence system also functional on their own elsewhere in the beetle’s body? Are there genetic differences between bombardier beetles and other Carabidae related to this system?
Without clear answers to these and other questions, the path of bombardier beetle evolution will remain an open and interesting question. However, biologists do not assume that these beetles did not evolve simply because the specific path has yet to be elucidated. And they certainly would not assume that an untestable, supernatural just-so story of cosmic proportions is the null hypothesis.
That, of course, is the difference between science and pseudoscience.
Larry Moran and PZ have both picked up on the term “DAP“. There have already been several more examples identified outside the realm of genomics in their comments threads. As such, I think that “DAP” can be given the following broadened definition:
Dog’s Ass Plot (DAP, or Dapper):
A graphical representation of data in any field that, through a lack of clear axis labels, selective inclusion/exclusion of data, visual presentation style, and/or other questionable characteristics, generates a misleading interpretation of the data in the viewer, especially by implying an illusory pattern that is not supported by the available data.
Can you indicate an example from your own subject of study? If so, please provide a link in the comments section. Bring on the Dappers!!
The word logodaedaly means “a capricious coinage of words”. It was coined by Plato in the 4th century BC (as “wordsmith”) and picked up by Ben Johnson in 1611 in its current English usage. That’s right, someone coined a term for the process of coining terms.
Sometimes new terms are very useful. Every profession has its own jargon, which for the most part helps experts to save time by having individual terms for specific items or ideas. On the other hand, the original meaning can be lost and the term can be badly misunderstood or misapplied when it moves from jargon to buzzword. “Junk DNA” is a case in point. Other terms may be coined to give a simple summary of a more complex idea. “The Onion Test” is an example: it’s not really about onions, but about providing a reminder that there is more diversity out there than one might otherwise have considered.
Finally, sometimes terms are coined just for fun. This is one of those times.
Several bloggers have drawn attention to the persistent assumption expressed by some authors that humans are the pinnacle of biological complexity, as reflected in certain graphical representations relating to non-coding DNA [Pharyngula, Sandwalk, Sunclipse, Genomicron]. Larry Moran’s discussion pointed to what must be the single worst figure of the genre, from an article in Scientific American. This figure forms the basis of a new term that I wish to coin.
Here is the figure in question:
In a previous post, I complained about the ridiculous division of groups (humans are vertebrates and vertebrates are chordates), the lack of labels on the X-axis, the ambiguous definition of “complexity” implied, and the blatant assumption, sans justification, that humans are the most complex organisms around.
I also noted the following issue:
The sloping of the bars within taxa suggests that this is meant to imply a relationship between genome size and complexity within groups as well, with the largest genomes (i.e., the most non-coding DNA) found in the most complex organisms. This would negate the goal of placing humans at the extreme, as our genome is average for a mammal and at the lower end of the vertebrate spectrum (some salamanders have 20x more DNA than humans). Indeed, the human datum would accurately be placed roughly below the dog’s ass in this figure if it included a proper sampling of diversity.
As a result, I hereby propose that all such figures, with unlabeled axes and clear yet unjustified assumptions about complexity, henceforth be dubbed “Dog’s Ass Plots”. “DAPs” or “Dappers” also are acceptable, as in “I’m surprised that the reviewers didn’t pick up on this DAP” or “Check out this figure, it’s a real Dapper”. (As an added bonus, “dapper” means “neat and trim” — which these figures certainly are; the problem is not that they don’t look slick, it’s that they are oversimplified). I have no doubt that plenty of examples can be found in subjects besides genomics, so please feel free to use it as needed in your own field.
It is commonly suggested by anti-evolutionists that recent discoveries of function in non-coding DNA support intelligent design and refute “Darwinism”. This misrepresents both the history and the science of this issue. I would like to provide some clarification of both aspects.
When people began estimating genome sizes (amounts of DNA per genome) in the late 1940s and early 1950s, they noticed that this is largely a constant trait within organisms and species. In other words, if you look at nuclei in different tissues within an organism or in different organisms from the same species, the amount of DNA per chromosome set is constant. (There are some interesting exceptions to this, but they were not really known at the time). This observed constancy in DNA amount was taken as evidence that DNA, rather than proteins, is the substance of inheritance.
These early researchers also noted that some “less complex” organisms (e.g., salamanders) possess far more DNA in their nuclei than “more complex” ones (e.g., mammals). This rendered the issue quite complex, because on the one hand DNA was thought to be constant because it’s what genes are made of, and yet the amount of DNA (“C-value”, for “constant”) did not correspond to assumptions about how many genes an organism should have. This (apparently) self-contradictory set of findings became known as the “C-value paradox” in 1971.
This “paradox” was solved with the discovery of non-coding DNA. Because most DNA in eukaryotes does not encode a protein, there is no longer a reason to expect C-value and gene number to be related. Not surprisingly, there was speculation about what role the “extra” DNA might be playing.
In 1972, Susumu Ohno coined the term “junk DNA“. The idea did not come from throwing his hands up and saying “we don’t know what it does so let’s just assume it is useless and call it junk”. He developed the idea based on knowledge about a mechanism by which non-coding DNA accumulates: the duplication and inactivation of genes. “Junk DNA,” as formulated by Ohno, referred to what we now call pseudogenes, which are non-functional from a protein-coding standpoint by definition. Nevertheless, a long list of possible functions for non-coding DNA continued to be proposed in the scientific literature.
In 1979, Gould and Lewontin published their classic “spandrels” paper (Proc. R. Soc. Lond. B 205: 581-598) in which they railed against the apparent tendency of biologists to attribute function to every feature of organisms. In the same vein, Doolittle and Sapienza published a paper in 1980 entitled “Selfish genes, the phenotype paradigm and genome evolution” (Nature 284: 601-603). In it, they argued that there was far too much emphasis on function at the organism level in explanations for the presence of so much non-coding DNA. Instead, they argued, self-replicating sequences (transposable elements) may be there simply because they are good at being there, independent of effects (let alone functions) at the organism level. Many biologists took their point seriously and began thinking about selection at two levels, within the genome and on organismal phenotypes. Meanwhile, functions for non-coding DNA continued to be postulated by other authors.
As the tools of molecular genetics grew increasingly powerful, there was a shift toward close examinations of protein-coding genes in some circles, and something of a divide emerged between researchers interested in particular sequences and others focusing on genome size and other large-scale features. This became apparent when technological advances allowed thoughts of sequencing the entire human genome: a question asked in all seriousness was whether the project should bother with the “junk”.
Of course, there is now a much greater link between genome sequencing and genome size research. For one, you need to know how much DNA is there just to get funding. More importantly, sequence analysis is shedding light on the types of non-coding DNA responsible for the differences in genome size, and non-coding DNA is proving to be at least as interesting as the genic portions.
To summarize,
Since the first discussions about DNA amount there have been scientists who argued that most non-coding DNA is functional, others who focused on mechanisms that could lead to more DNA in the absence of function, and yet others who took a position somewhere in the middle. This is still the situation now.
Lots of mechanisms are known that can increase the amount of DNA in a genome: gene duplication and pseudogenization, duplicative transposition, replication slippage, unequal crossing-over, aneuploidy, and polyploidy. By themselves, these could lead to increases in DNA content independent of benefits for the organism, or even despite small detrimental impacts, which is why non-function is a reasonable null hypothesis.
Evidence currently available suggests that about 5% of the human genome is functional. The least conservative guesses put the possible total at about 20%. The human genome is mid-sized for an animal, which means that most likely a smaller percentage than this is functional in other genomes. None of the discoveries suggest that all (or even more than a minor percentage) of non-coding DNA is functional, and the corollary is that there is indirect evidence that most of it is not.
Identification of function is done by evolutionary biologists and genome researchers using an explicit evolutionary framework. One of the best indications of function that we have for non-coding DNA is to find parts of it conserved among species. This suggests that changes to the sequence have been selected against over long stretches of time because those regions play a significant role. Obviously you can not talk about evolutionarily conserved DNA without evolutionary change.
Examples of transposable elements acquiring function represent co-option. This is the same phenomenon that is involved in the evolution of complex features like eyes and flagella. In particular, co-option of TEs appears to have happened in the evolution of the vertebrate immune system. Again, this makes no sense in the absence of an evolutionary scenario.
Most transposable elements do not appear to be functional at the organism level. In humans, most are inactive molecular fossils. Some are active, however, and can cause all manner of diseases through their insertions. To repeat: some transposons are functional, some are clearly deleterious, and most probably remain more or less neutral.
Any suggestions that all non-coding DNA is functional must explain why an onion needs five times more of it than you do. So far, none of the proposed unilateral functions has done this. It therefore remains most reasonable to take a pluralistic approach in which only some non-coding elements are functional for organisms.
I realize that this will have no effect on the arguments made by anti-evolutionists, but I hope it at least clarifies the issue for readers who are interested in the actual science involved and its historical development.
The results of the proof-of-principle phase of ENCODE, the Encyclopedia of DNA Elements Project, appear in the June 14 issue of Nature. It’s a very interesting project, and it has revealed a few more surprises (or at least, added evidence in favour of previously surprising observations). I will probably post more about it soon, but for the time being let me just offer a brief apology to the science writers out there whom I have given a hard time about invoking sloppy language to describe non-coding DNA, sequencing, and genomes (recent example, but one I will leave alone, ‘Junk’ DNA makes compulsive reading online at New Scientist).
The reason I am sorry is that I simply cannot hold you to a higher standard than is maintained by one of the most prestigious journals on planet Earth. You see, Nature has decided to depict the ENCODE project on the cover as “Decoding the Blueprint”. Needless to say (again), genomes are not blueprints (as the ENCODE project shows!) and no one is decoding anything at this point.
I have saidallthisbefore, and even I am getting tired of my complaints about it. Thus, I will focus only on the interesting science in a later post.
This is a very quick post about something that is annoying to many biologists — the misuse of species names. It is prompted by a news headline I just saw, which is about the 106th time I have seen this problem (“T. Rex”).
The binomial naming system was developed by Linnaeus in the 18th century and is still in use in modern biology. It consists of two names for each species — or, more specifically, a genus and species name. Every species should have only one genus/species name, although it takes work to correct multiple names for the same species (synonyms) and to split different species that are grouped together into a single name (cryptic species). The genus is a broader category and is (usually) comprised of many species. I will not get into the difficulty in defining what species are (though this is an interesting issue), only a few points about the use of terminology.
The plural of “species” is “species”. The singular of “species” is also “species”, not “specie”. “Specie” refers to coins.
The plural of “genus” is “genera”.
Genus and species names are always written in italics.
Genus names are capitalized, species designations are not. For example, it should be “Tyrannosaurus rex“, not “Tyrannosaurus Rex“.
Once you have defined the species name (or if it is very well known), you can abbreviate the genus. For example, “T. rex“. The same capitalized/lowercase and italics rules apply (it does not become “T. Rex“, though watch for automated spell checkers to un-correct this for you).
Some species are further partitioned into subspecies (although this is a more nebulous category than species); subspecies designations follow the same rules as species names (e.g., “Canis lupus familiaris“).
Humans are categorized as Homo sapiens, the name given by Linnaeus in 1758. It means “man, the wise”. Homo sapiens is a proper noun and not a common noun, such that one human is not a “Homo sapien”.
The following is a re-post of my comments on the recently postedNoncoding DNA and Junk DNA at Sandwalk. Needless to say, I am quite pleased to see such active discussion about non-coding DNA. Passages in italics are excerpts from the original article.
TR Gregory said…
Ryan Gregory has serious doubts about the usefulness of the term as he explains in his excellent article A word about “junk DNA”.
Just to clarify, I think the term could be useful — indeed, it was useful when Ohno coined it. The problem is that it is seldom used in an appropriate way. If the meaning were specified explicitly to be “regions strongly suspected of being non-functional with evidence to back it up” (which, incidentally, is not the original definition according to Ohno (1972) or Comings (1972)), and if people used it only in this way, then I would not have a problem with this. But given the difficulty that people seem to have in accepting that some DNA may truly not have a function at the organism level, I don’t know if we could ever get it to be used with such precision.
…a new term, Junctional DNA, to describe DNA that probably has a function but that function isn’t known… think we don’t need to go there. It’s sufficient to remind people that lots of DNA outside of genes has a function and these functions have been known for decades.
That neologism was suggested in response to Minkel’s appeal for a term that would “make the distinction between functional and nonfunctional noncoding DNA clear to a popular audience”. My main suggestion was to call DNA by what it is known to be, if at all possible, by function (“regulatory DNA”, “structural DNA”) or by type (“pseudogene”, “transposable element”, “intron”). Your definition of “junk DNA” is also more precise than most usages, meaning that you specify that the term only be applied to sequences for which there is evidence (not just assumption) of non-function. That leaves us with something in between for journalists to talk about with a catchy buzzword. “Junctional DNA” lets them specify that we’re not talking about “junk DNA” or “functional DNA” — i.e., there is some evidence for function (e.g., being conserved) but no evidence of what that function is. The main utility would be to stop the very frustrating leap that gets made from “this 1% of the genome may have a function, so the whole thing must have this function” kind of reporting. Now they could say “another 1% has moved into the category of ‘junctional DNA'”. I think that would be considerably less misleading than current wording.
Note that I’m avoiding the term “noncoding” DNA here. This is because to me the term “coding DNA” only refers to the coding region of a gene that encodes a protein … there are many genes for RNAs that are not properly called coding regions so they would fall into the noncoding DNA category … introns in eukaryotic genomes would be “noncoding DNA” as far as I’m concerned. I think that Ryan Gregory and others use the term “noncoding DNA” to refer to all DNA that’s not part of a gene instead of all DNA that’s not part of the coding region of a protein encoding gene. I’m not certain of this.
By definition, non-coding DNA is, and always has been, everything other than exons. The reason this is relevant is that early work in genome biology assumed that there should be a 1 to 1 correspondence between DNA content and protein-coding gene number. This is work that occurred for at least two decades before the discovery of introns, pseudogenes, and other non-coding DNA. Now we have more descriptive names for the categories of DNA that are not the genes, all the genes, and nothing but the genes. I actually don’t know of anyone else who would have a problem calling introns, pseudogenes, and regulatory regions “non-coding DNA”. Certainly, Ohno, Crick, and many others have historically put introns in the same non-protein-coding grouping as pseudogenes. It’s just a category — you also have more specific subcategories to apply to each of the types of non-coding DNA. Perhaps your objection relates to an undue emphasis on the distinction between exons and everything else — well, that’s the history of the past half century of this field, so it should be no surprise that the terminology reflects this.
Read Gregory’s article for the short concise version of this dispute. What it means is that junk DNA threatens the worldviews of both Dembski and Dawkins!
Not quite. What you’re leaving out of this is the possibility of multiple levels of selection. In the original edition of The Selfish Gene (1976, p.76), Dawkins argued that “the simplest way to explain the surplus DNA is to suppose that it is a parasite, or at best a harmless but useless passenger, hitching a ride in the survival machines created by the other DNA”. Cavalier-Smith (1977) drew a similar conclusion (before he had read Dawkins), and Doolittle and Sapienza (1980) and Orgel and Crick (1980) [yes, that Crick] independently developed the concept of “selfish DNA” a few years later. This is an explicitly multi-level selection approach because it specifies that non-coding DNA can be present due to selection within the genome rather than exclusively on the organism (or gene, in Dawkins’s case) (see, e.g., Gregory 2004, 2005). (Incidentally, this idea of parasitic DNA dates back at least to 1945, when Gunnar Östergren characterized B chromosomes in this fashion). Of course, they tended to do what Ohno did and applied this one idea to all non-coding DNA, which is too ambitious. The modern view is more pluralistic (see, e.g., Pagel and Johnstone 1992 vs. Gregory 2003). Some non-coding DNA is just accumulated “junk” (in the definition of evidence-supported non-function that you espouse). Some (perhaps most) is “selfish” or “parasitic” and persists because there is selection within the genome as well as on organisms (in fact, an argument could be, and has been, made that “selfish DNA” would be a much more accurate term than “junk DNA” for most non-coding DNA). Some non-coding DNA is clearly functional at the organism level, including regulatory regions and chromosome structure components. Some of these latter functional non-coding DNA sequences are derived from elements that originally were of one of the first two types, most notably transposable elements that take on a regulatory function through co-option (or, in another manner of thinking, that undergo a shift in level of selection).
Junk DNA is not noncoding DNA and anyone who claims otherwise just doesn’t know what they’re talking about.
I’m afraid I don’t follow what you mean here. By your definition, “junk DNA” is any non-functional sequence of DNA, including pseudogenes (i.e., the original meaning). Those sequences do not encode proteins. Hence, your version of junk DNA is non-coding. I think this reflects the confusion that is imposed by the term “junk DNA”, which is why I generally think it is more obfuscating than enlightening.
________
References
Cavalier-Smith, T. 1977. Visualising jumping genes. Nature 270: 10-12.
Comings, D.E. 1972. The structure and function of chromatin. Advances in Human Genetics 3: 237-431.
Dawkins, R. 1976. The Selfish Gene. Oxford University Press, Oxford.
Doolittle, W.F. and C. Sapienza. 1980. Selfish genes, the phenotype paradigm and genome evolution. Nature 284: 601-603.
Gregory, T.R. 2003. Variation across amphibian species in the size of the nuclear genome supports a pluralistic, hierarchical approach to the C-value enigma. Biological Journal of the Linnean Society 79: 329-339.
Gregory, T.R. 2004. Macroevolution, hierarchy theory, and the C-value enigma. Paleobiology 30: 179-202.
Gregory, T.R. 2005. Macroevolution and the genome. In The Evolution of the Genome (ed. T.R. Gregory), pp. 679-729. Elsevier, San Diego.
Ohno, S. 1972. So much “junk” DNA in our genome. In Evolution of Genetic Systems (ed. H.H. Smith), pp. 366-370. Gordon and Breach, New York.
Orgel, L.E. and F.H.C. Crick. 1980. Selfish DNA: the ultimate parasite. Nature 284: 604-607.
Östergren, G. 1945. Parasitic nature of extra fragment chromosomes. Botaniska Notiser 2: 157-163.
Pagel, M. and R.A. Johnstone. 1992. Variation across species in the size of the nuclear genome supports the junk-DNA explanantion for the C-value paradox. Proceedings of the Royal Society of London, Series B: Biological Sciences 249: 119-124.

 Sometimes it is helpful to have a catchy word to describe one’s type of research. I think that’s why “omics” words are so popular — they encapsulate a complex combination of approaches (usually something + genomics, or something-more-than-genomics) in a memorable way that immediately conveys the gist of the field. “Metagenomics” is a good example — it’s the study of a larger assemblage of genomes than just one, usually from an environmental sample of microbes. “Proteiomics” is another, or “transcriptomics”. Of course, this can get out of hand (see here). However, I think the study of genome size (which predates molecular genetics, let alone genomics) deserves a catchy moniker. The problem is, I haven’t really come up with one in the past, so I just end up saying “I study the total amount of DNA in different species of animals, which includes genes and all the other sequences, most of which are non-coding and…” — well, you get the idea.
Sometimes it is helpful to have a catchy word to describe one’s type of research. I think that’s why “omics” words are so popular — they encapsulate a complex combination of approaches (usually something + genomics, or something-more-than-genomics) in a memorable way that immediately conveys the gist of the field. “Metagenomics” is a good example — it’s the study of a larger assemblage of genomes than just one, usually from an environmental sample of microbes. “Proteiomics” is another, or “transcriptomics”. Of course, this can get out of hand (see here). However, I think the study of genome size (which predates molecular genetics, let alone genomics) deserves a catchy moniker. The problem is, I haven’t really come up with one in the past, so I just end up saying “I study the total amount of DNA in different species of animals, which includes genes and all the other sequences, most of which are non-coding and…” — well, you get the idea.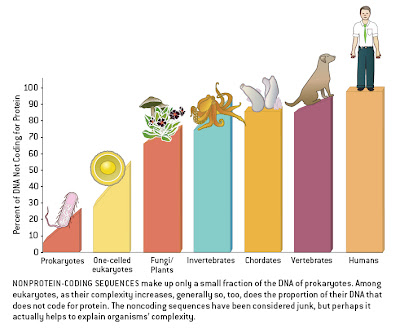

Ryan Gregory has serious doubts about the usefulness of the term as he explains in his excellent article A word about “junk DNA”.
Just to clarify, I think the term could be useful — indeed, it was useful when Ohno coined it. The problem is that it is seldom used in an appropriate way. If the meaning were specified explicitly to be “regions strongly suspected of being non-functional with evidence to back it up” (which, incidentally, is not the original definition according to Ohno (1972) or Comings (1972)), and if people used it only in this way, then I would not have a problem with this. But given the difficulty that people seem to have in accepting that some DNA may truly not have a function at the organism level, I don’t know if we could ever get it to be used with such precision.
…a new term, Junctional DNA, to describe DNA that probably has a function but that function isn’t known… think we don’t need to go there. It’s sufficient to remind people that lots of DNA outside of genes has a function and these functions have been known for decades.
That neologism was suggested in response to Minkel’s appeal for a term that would “make the distinction between functional and nonfunctional noncoding DNA clear to a popular audience”. My main suggestion was to call DNA by what it is known to be, if at all possible, by function (“regulatory DNA”, “structural DNA”) or by type (“pseudogene”, “transposable element”, “intron”). Your definition of “junk DNA” is also more precise than most usages, meaning that you specify that the term only be applied to sequences for which there is evidence (not just assumption) of non-function. That leaves us with something in between for journalists to talk about with a catchy buzzword. “Junctional DNA” lets them specify that we’re not talking about “junk DNA” or “functional DNA” — i.e., there is some evidence for function (e.g., being conserved) but no evidence of what that function is. The main utility would be to stop the very frustrating leap that gets made from “this 1% of the genome may have a function, so the whole thing must have this function” kind of reporting. Now they could say “another 1% has moved into the category of ‘junctional DNA'”. I think that would be considerably less misleading than current wording.
Note that I’m avoiding the term “noncoding” DNA here. This is because to me the term “coding DNA” only refers to the coding region of a gene that encodes a protein … there are many genes for RNAs that are not properly called coding regions so they would fall into the noncoding DNA category … introns in eukaryotic genomes would be “noncoding DNA” as far as I’m concerned. I think that Ryan Gregory and others use the term “noncoding DNA” to refer to all DNA that’s not part of a gene instead of all DNA that’s not part of the coding region of a protein encoding gene. I’m not certain of this.
By definition, non-coding DNA is, and always has been, everything other than exons. The reason this is relevant is that early work in genome biology assumed that there should be a 1 to 1 correspondence between DNA content and protein-coding gene number. This is work that occurred for at least two decades before the discovery of introns, pseudogenes, and other non-coding DNA. Now we have more descriptive names for the categories of DNA that are not the genes, all the genes, and nothing but the genes. I actually don’t know of anyone else who would have a problem calling introns, pseudogenes, and regulatory regions “non-coding DNA”. Certainly, Ohno, Crick, and many others have historically put introns in the same non-protein-coding grouping as pseudogenes. It’s just a category — you also have more specific subcategories to apply to each of the types of non-coding DNA. Perhaps your objection relates to an undue emphasis on the distinction between exons and everything else — well, that’s the history of the past half century of this field, so it should be no surprise that the terminology reflects this.
Read Gregory’s article for the short concise version of this dispute. What it means is that junk DNA threatens the worldviews of both Dembski and Dawkins!
Not quite. What you’re leaving out of this is the possibility of multiple levels of selection. In the original edition of The Selfish Gene (1976, p.76), Dawkins argued that “the simplest way to explain the surplus DNA is to suppose that it is a parasite, or at best a harmless but useless passenger, hitching a ride in the survival machines created by the other DNA”. Cavalier-Smith (1977) drew a similar conclusion (before he had read Dawkins), and Doolittle and Sapienza (1980) and Orgel and Crick (1980) [yes, that Crick] independently developed the concept of “selfish DNA” a few years later. This is an explicitly multi-level selection approach because it specifies that non-coding DNA can be present due to selection within the genome rather than exclusively on the organism (or gene, in Dawkins’s case) (see, e.g., Gregory 2004, 2005). (Incidentally, this idea of parasitic DNA dates back at least to 1945, when Gunnar Östergren characterized B chromosomes in this fashion). Of course, they tended to do what Ohno did and applied this one idea to all non-coding DNA, which is too ambitious. The modern view is more pluralistic (see, e.g., Pagel and Johnstone 1992 vs. Gregory 2003). Some non-coding DNA is just accumulated “junk” (in the definition of evidence-supported non-function that you espouse). Some (perhaps most) is “selfish” or “parasitic” and persists because there is selection within the genome as well as on organisms (in fact, an argument could be, and has been, made that “selfish DNA” would be a much more accurate term than “junk DNA” for most non-coding DNA). Some non-coding DNA is clearly functional at the organism level, including regulatory regions and chromosome structure components. Some of these latter functional non-coding DNA sequences are derived from elements that originally were of one of the first two types, most notably transposable elements that take on a regulatory function through co-option (or, in another manner of thinking, that undergo a shift in level of selection).
Junk DNA is not noncoding DNA and anyone who claims otherwise just doesn’t know what they’re talking about.
I’m afraid I don’t follow what you mean here. By your definition, “junk DNA” is any non-functional sequence of DNA, including pseudogenes (i.e., the original meaning). Those sequences do not encode proteins. Hence, your version of junk DNA is non-coding. I think this reflects the confusion that is imposed by the term “junk DNA”, which is why I generally think it is more obfuscating than enlightening.
________
References
Cavalier-Smith, T. 1977. Visualising jumping genes. Nature 270: 10-12.
Comings, D.E. 1972. The structure and function of chromatin. Advances in Human Genetics 3: 237-431.
Dawkins, R. 1976. The Selfish Gene. Oxford University Press, Oxford.
Doolittle, W.F. and C. Sapienza. 1980. Selfish genes, the phenotype paradigm and genome evolution. Nature 284: 601-603.
Gregory, T.R. 2003. Variation across amphibian species in the size of the nuclear genome supports a pluralistic, hierarchical approach to the C-value enigma. Biological Journal of the Linnean Society 79: 329-339.
Gregory, T.R. 2004. Macroevolution, hierarchy theory, and the C-value enigma. Paleobiology 30: 179-202.
Gregory, T.R. 2005. Macroevolution and the genome. In The Evolution of the Genome (ed. T.R. Gregory), pp. 679-729. Elsevier, San Diego.
Ohno, S. 1972. So much “junk” DNA in our genome. In Evolution of Genetic Systems (ed. H.H. Smith), pp. 366-370. Gordon and Breach, New York.
Orgel, L.E. and F.H.C. Crick. 1980. Selfish DNA: the ultimate parasite. Nature 284: 604-607.
Östergren, G. 1945. Parasitic nature of extra fragment chromosomes. Botaniska Notiser 2: 157-163.
Pagel, M. and R.A. Johnstone. 1992. Variation across species in the size of the nuclear genome supports the junk-DNA explanantion for the C-value paradox. Proceedings of the Royal Society of London, Series B: Biological Sciences 249: 119-124.